Analyse des obligations SS
Si la séquence génétique qui code pour l'anticorps cible indique un résidu cystéine, les lignes directrices exigent la détermination du nombre et des positions de tous les groupes sulfhydryle libres et des liaisons disulfure. Ceci est évalué par cartographie peptidique (dans des conditions réductrices et non réductrices), par spectrométrie de masse ou par une autre méthode appropriée.
L’analyse présentée ici utilise une combinaison de cartographie peptidique et de spectrométrie de masse. Cette méthode consiste à digérer la protéine cible dans une enzyme protéolytique telle que la trypsine dans diverses conditions et à comparer les poids moléculaires des fragments peptidiques générés avec les valeurs théoriques. Les procédures de test et les résultats pour l'analyse du lysozyme humain sont présentés.
Flux d'expérimentation et de recherche
- Utilisez le kit de synthèse de protéines acellulaires (cellule d’insecte Transdirect) pour synthétiser le lysozyme humain dans un tube à essai. (La figure 1 montre la séquence du lysozyme humain synthétisé.)
- Purifier dans une colonne d'affinité (Strep-Tactin super flow, fabriqué par QIAGEN, Allemagne).
- Dénaturer à l'aide d'urée 8M.
- Digestion par la trypsine après trois types de prétraitement : non traité (conservation de la liaison SS) ; S-alkylation (alkylation de tous les groupes SH libres existants) ; réduction et alkylation (réduction des liaisons SS suivie d'une alkylation des groupes SH)
Résultats
La figure 1 montre la séquence d'acides aminés et les liaisons SS prédites (indiquées par des lignes droites) du lysozyme humain synthétisé dans le tube à essai. Le tableau 1 montre les valeurs théoriques et les valeurs mesurées MS pour les fragments peptidiques digérés par la trypsine (associés aux liaisons SS).
La figure 2 montre une partie du spectre MS des fragments peptidiques digérés par la trypsine.
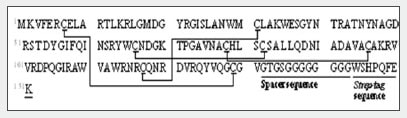
Fig. 1 Séquence d'acides aminés et liaisons SS prévues du lysozyme humain synthétisé
L'ion m/z 3239,22 disparaît en raison de la réduction et de l'alkylation du fragment peptidique B, indiquant que le peptide 3 et le peptide 4 forment des liaisons SS intramoléculaires (Fig. 2). La même méthode a confirmé les liaisons SS intramoléculaires dans le fragment peptidique C. (Données omises.)
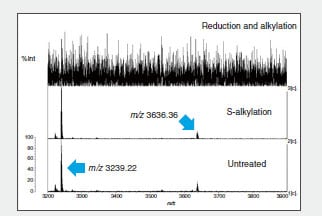
Fig. 2 Spectre MS des fragments peptidiques récupérés (m/z 3 200 à m/z 3 900)
La comparaison des spectres de réduction/alkylation et de MS non traité pour le fragment peptidique A indique que le peptide 1 et
le peptide 2 forment des liaisons SS intramoléculaires. Cependant, la combinaison avec le résidu cystéine ne peut pas être déterminée
uniquement à partir des résultats de la figure 2.
Pour confirmer cette combinaison, les liaisons SS intramoléculaires du peptide 1 ont été identifiées par LC pour séparer et purifier
le fragment peptidique A, par digestion dans une autre protéase (thermolysine) et par détermination de la masse des
fragments générés (Fig. 3).
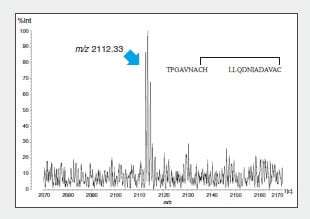
Fig. 3 Spectre MS du digestat de thermolysine du fragment peptidique A (m/z 3636,36)
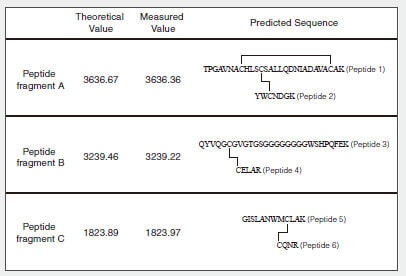
Tableau 1 Valeurs théoriques et valeurs mesurées pour les fragments peptidiques prédits (associés aux liaisons SS)
AXIMA Performance MALDI-TOF MS

L'AXIMA Performance est l'un des outils les plus puissants en spectrométrie de masse, fournissant des spectres riches en informations avec une plus grande sensibilité et une plus grande confiance dans l'identification . Il s'agit d'un système TOF-TOF extrêmement polyvalent et puissant, intégrant des flux de travail pour un large éventail de besoins analytiques.
- De la MS/MS à haute énergie de la protéomique et d'autres échantillons biologiques et organiques à l'analyse sans compromis de protéines intactes de masse élevée
- Véritable MS/MS - CID à haute énergie avec une énergie de collision de cadre en laboratoire de 20 keV
- Résolution optimale de sélection des ions précurseurs grâce à une technologie de déclenchement révolutionnaire
- Sensibilité exceptionnelle - conception sans compromis, pour garantir qu'aucun signal MS/MS n'est rejeté